在生物制藥領(lǐng)域,無菌過濾工藝是確保最終產(chǎn)品質(zhì)量和安全性的關(guān)鍵環(huán)節(jié)之一。其中,用于去除細(xì)菌和控制生物負(fù)荷的除菌過濾系統(tǒng)一直是無菌過濾工藝中的重要環(huán)節(jié)。
PUPSIT定義
根據(jù)EU 新版 GMP 的相關(guān)規(guī)定“8.87 The integrity of the sterilised filter assembly should be verified by integrity testing before use (pre-use post sterilisation integrity test or PUPSIT), to check for damage and loss of integrity caused by the filter preparation prior to use.”
PUPSIT,即“使用前、滅菌后完整性測試”(Pre-Use Post-Sterilization Integrity Test),是無菌制藥工藝中的一項(xiàng)重要測試措施。
由于滅菌過程可能會對過濾器產(chǎn)生影響,如導(dǎo)致微小破損、孔徑變化,或引入“掩蓋缺陷”的風(fēng)險(xiǎn),PUPSIT幫助制藥企業(yè)在生產(chǎn)啟動之前確認(rèn)過濾器是否能有效地過濾微生物和其他顆粒,確保產(chǎn)品的最終無菌性。
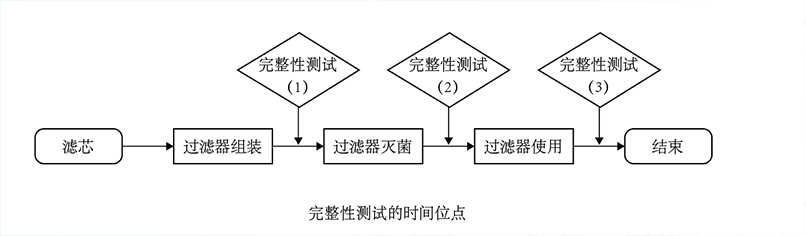
實(shí)施要點(diǎn)
1. 測試時機(jī)與流程設(shè)計(jì)
嚴(yán)格時序:必須在過濾器完成滅菌(如濕熱滅菌、輻照滅菌)后,且在實(shí)際使用(如藥液過濾、無菌工藝)前進(jìn)行,避免滅菌后二次污染或操作損傷。
無縫銜接:若滅菌后無法立即測試(如過濾器已安裝于系統(tǒng)),需確保測試前系統(tǒng)處于密閉狀態(tài),防止外界污染。
2. 滅菌方法兼容性
濕熱滅菌(SIP):需驗(yàn)證高溫高壓對濾膜(如PES、PVDF)的影響,避免膜孔徑變形或密封件老化。
輻照滅菌:注意γ射線或電子束可能降低濾膜機(jī)械強(qiáng)度,需通過完整性測試確認(rèn)無損傷。
環(huán)氧乙烷(EO)滅菌:需徹底解析殘留氣體,避免影響測試結(jié)果(如擴(kuò)散流測試干擾)。
3. 完整性測試方法選擇
起泡點(diǎn)測試(Bubble Point):適用于膜孔徑較大(≥0.2μm)的除菌過濾器,需設(shè)定滅菌前后的閾值(如0.2μm濾膜起泡點(diǎn)≥3.5 bar)。
擴(kuò)散流/前進(jìn)流測試(Diffusion Flow):更敏感,適用于小孔徑或多層濾膜,需根據(jù)濾膜材質(zhì)和滅菌條件校準(zhǔn)標(biāo)準(zhǔn)值。
壓力衰減測試(Pressure Hold):適用于在線測試,需確保系統(tǒng)密閉性(如管路、接頭無泄漏)。
4. 測試系統(tǒng)設(shè)計(jì)
在線測試 vs. 離線測試:
在線測試:直接在生產(chǎn)系統(tǒng)中進(jìn)行,需驗(yàn)證測試設(shè)備(如完整性測試儀)與工藝管路的兼容性。
離線測試:適用于小型或獨(dú)立過濾器,需確保拆卸-測試-重新安裝過程的無菌性。
自動化集成:優(yōu)先采用自動化測試設(shè)備,減少人為操作誤差,確保數(shù)據(jù)完整性(如電子記錄符合ALCOA+原則)。
5. 驗(yàn)證與接受標(biāo)準(zhǔn)
滅菌前后對比:需建立滅菌前(出廠)與滅菌后(PUPSIT)的完整性測試數(shù)據(jù)基線,確認(rèn)滅菌過程未導(dǎo)致性能漂移。
接受標(biāo)準(zhǔn):依據(jù)過濾器廠家標(biāo)定值,結(jié)合工藝驗(yàn)證數(shù)據(jù)設(shè)定(如擴(kuò)散流≤10 mL/min·cm2)。
失敗處理:若測試失敗,需啟動偏差調(diào)查(是否為滅菌損傷、安裝不當(dāng)或測試誤差),并更換過濾器。
風(fēng)險(xiǎn)評估與關(guān)鍵控制
風(fēng)險(xiǎn)點(diǎn):滅菌參數(shù)超限(如溫度過高)、過濾器安裝不當(dāng)(如O型圈損壞)、測試環(huán)境(如非潔凈區(qū)操作)。
控制措施:通過工藝驗(yàn)證(如滅菌循環(huán)驗(yàn)證)、操作SOP培訓(xùn)、環(huán)境監(jiān)控(如粒子計(jì)數(shù))降低風(fēng)險(xiǎn)。
法規(guī)與文件要求
GMP/FDA合規(guī):符合FDA《無菌工藝指南》、EU GMP Annex 1(2022)要求,明確PUPSIT為強(qiáng)制步驟。
?歐盟GMP附錄1:2022年發(fā)布的新版附錄1明確要求,在沒有顯著工藝限制(如溶液體積較小)的情況下,制藥企業(yè)必須在使用前對滅菌后的除菌過濾器進(jìn)行PUPSIT測試。這不僅是為了防止微生物污染,還因滅菌過程可能導(dǎo)致濾器的完整性下降,從而影響藥品的無菌質(zhì)量。
?美國FDA:FDA的相關(guān)指南雖然未明確要求所有情況下必須執(zhí)行PUPSIT,但強(qiáng)調(diào)根據(jù)產(chǎn)品特性進(jìn)行風(fēng)險(xiǎn)評估,并在必要時執(zhí)行完整性檢測。對于那些無法在滅菌后實(shí)施PUPSIT的產(chǎn)品,需基于科學(xué)依據(jù)采取其他風(fēng)險(xiǎn)控制措施。
?WHO和PIC/S:全球化的GMP指南如WHO和PIC/S同樣支持PUPSIT,特別是針對出口或跨國銷售的無菌藥品,要求嚴(yán)格執(zhí)行PUPSIT的流程,以確保最終產(chǎn)品質(zhì)量達(dá)到國際標(biāo)準(zhǔn)。

文件記錄:需完整記錄滅菌參數(shù)、測試結(jié)果、操作人員、設(shè)備編號等信息,支持產(chǎn)品批次放行。
審計(jì)重點(diǎn):檢查測試數(shù)據(jù)趨勢分析、偏差記錄、過濾器更換頻率是否合理。
典型案例應(yīng)用
注射液除菌過濾:在線PUPSIT確保藥液過濾前過濾器完整性。
生物反應(yīng)器進(jìn)氣過濾器:滅菌后測試防止細(xì)胞培養(yǎng)污染。
無菌包裝器械:測試呼吸袋過濾器是否在EO滅菌后仍維持無菌屏障。
總結(jié)
PUPSIT是保障無菌工藝安全的核心環(huán)節(jié),需通過科學(xué)的驗(yàn)證、嚴(yán)格的流程控制和合規(guī)的文件管理實(shí)現(xiàn)。實(shí)施時應(yīng)結(jié)合具體工藝特點(diǎn)(如滅菌方式、過濾器類型),確保測試結(jié)果的可靠性與重現(xiàn)性。

 英文
英文  西語
西語 阿拉伯語
阿拉伯語 日語
日語
